Monte Carlo Simulations
 @Sudheer Ganisetti
@Sudheer Ganisetti
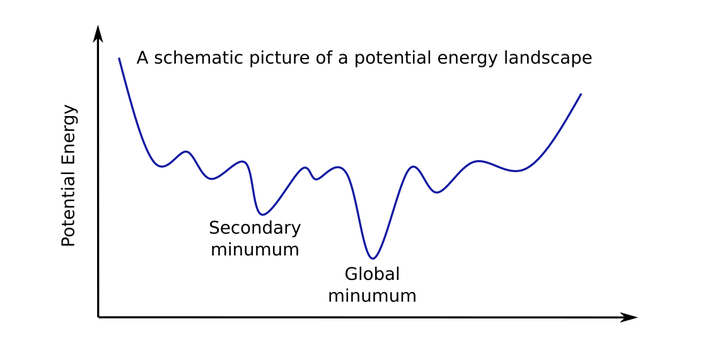
Monte Carlo method was used to tackle the global optimization problem of molecular structure. A Fortran code was developed to optimize a 13 atom molecular structure. Molecular simulations are very convenient to get insight into the structure and thermal behavior of small atomic clusters. Atomic structures are finite size aggregates containing between a few and millions of particles. Due to their small dimensions, their theoretical study faces serious difficulties. The most properties exhibit a strong dependence on the number of atoms. These difficulties actually begin with the unoptimized structure, from which other characteristics will develop. Predicting the temperature where the cluster changes phase also requires simulations. By the use of numerical simulation method, we can achieve phase transition easily. Metropolis Monte Carlo method was chosen to sample the configuration space for structural optimization, and for simulating thermal equilibrium in a small Lennard-Jones cluster. Simulated Annealing technique stands as one of the most general and efficient ways of addressing the global optimization problem. Simulated annealing essentially consists of cooling slowly. The initial temperature, cooling schedule and total simulation length are the few but important parameters of a simulated annealing optimization. Starting from a random atomic configuration, the MC simulation is carried out following the metropolis acceptance rule, and after some number of steps the temperature was decreased. In this way, the most stable configuration of a 13 atom cluster was found by energy minimization.